Zombie cyclophilins catalyze HIV capsid isomerization

If you're going to study the role an enzyme plays in a biological pathway, it's often useful to "kill" it with a mutation. For example, the proline
cis-
trans isomerase cyclophilin A (CypA) needs a particular arginine residue for its chemistry, so mutations that remove or alter that functional group, like R55K and R55A, should destroy the protein's function and have effects on the related pathways that help illustrate its role. The hydrophobic pocket it uses to bind substrates is made by residues like H126, F113, and W121. Growing or shrinking those residues should alter the shape of the pocket and change binding or activity, leaving the enzyme "dead".
Using model reactions and various binding assays, researchers have previously examined a number of these mutants (4,7) and found that they diminish isomerase activity and alter inhibition. However, a detailed study of the effects of the mutations on CypA's catalytic cycle has not been performed. Former Kern lab members Daryl Bosco (now a professor at UMass Medical) and Elan Eisenmesser (now at UCHSC) examined these mutants in greater detail to see how they really behaved. I also contributed some data at the last minute, when the third reviewer requested we study an additional mutant, prompting a scene that I promise was not
too much like that
Downfall parody. In every case we found that these enzymes, although significantly impaired, weren't as dead as they had seemed.
You had me at "dunno"

CAN bound to CypA, from PDB structure
1AK4 (5).
CypA residues are labeled in black, CAN residues in red.One key aspect of this work is that it involves a physiological substrate of CypA, namely the N-terminal domain of the HIV-1 capsid protein (CAN). Mature HIV-1 virions contain CypA that is bound to proline 90 of CAN. The absence of CypA dramatically reduces their ability to infect their target cells, which we know from experiments with mutant CA proteins as well as ones involving the CypA inhibitor cyclosporin (3). What we don't know about the system is exactly what CypA does for HIV-1. The crystal structure (right) of CAN in complex with CypA appears to only capture the
trans isomer configuration (5), but
for reasons I have discussed previously on this blog, that's not particularly informative. We know, largely from Daryl and Elan's previous research on the system (2), that when CAN is floating free in solution CypA will catalyze isomerization, but in the context of a fully assembled capsid that situation could conceivably change.
This leaves us with three possibilities for CypA's function in the capsid. Catalysis of
cis-
trans isomerization of the proline bond could be important. Or, maybe all capsid needs is for CypA to bind at P90, and catalysis is irrelevant. And perhaps neither of these functions matters and CypA just needs to be hanging around for some other reason. To address these possibilities, Saphire
et al. performed an elegant series of experiments where they sneaked an engineered CypA protein into another part of the capsid by fusing it to a protein called Vpr. When they replaced the normal CypA sequence with a mutant (H126A) that was supposed to abrogate both binding and catalysis, HIV-1 could still infect CD4+ cells (6). But, how sure can we be that H126A, or any other mutant, is actually "dead"?
You can't measure what you can't see
The problem with proline isomerization, from a biochemist's standpoint, is that it's a difficult reaction to detect. While switching between isomerization states may have structurally significant effects, there's no direct spectroscopic signal to tell you whether a proline bond is in the
cis or
trans conformation. Even if there was, most proteins have many prolines and so the signal of the bond you care about might be difficult to separate from the bonds you don't.
You can get around this difficulty using a coupled reaction with a model substrate. CypA catalyzes the isomerization of tetrapeptides of the form AXPF pretty efficiently. As it turns out, sequences like this are also good substrates for the protease chymotrypsin, but there's a catch. Chymotrypsin only cleaves substrates where the proline bond is in the
trans configuration. So, what you can do is take a substrate like succinyl-Ala-Ala-Pro-Phe-p-nitroanilide, add a tiny amount of cyclophilin, and then dump in a huge amount of chymotrypsin. With enough chymotrypsin, the peptide that's already
trans will be cleaved before the solution stabilizes, causing a color change (due to the pNA) that can be measured with a conventional spectrophotometer. Then you can monitor the conversion of the remaining substrate from
cis to
trans, because there's so much chymotrypsin that cleavage after isomerization is essentially instantaneous.
This works reasonably well, but it has some limitations. You're stuck with a model peptide that may not behave very much like your particular protein substrate. You're only following the
cis-to-
trans reaction, and even that comes with limited detail. Also, performing the experiment takes some careful work, because if you add too much of your CypA the reaction will end before the solution turbulence settles, and if you add too little, the intrinsic
cis-
trans isomerization will interfere with your catalytic measurement.
Although proline isomerization is a difficult reaction to follow by spectrophotometry, it's actually quite convenient to assay by NMR. Because CypA catalyzes the reaction in both directions, it's impossible to exhaust the substrate. The kinetics can therefore be measured at equilibrium using NOESY and ZZ-exchange experiments (2). Of course the experiment is limited by our ability to express isotopically-labeled substrate proteins, but provided we can do that and visualize the active site in our spectra, then we can observe catalysis of the native substrate. When you perform this experiment on these various "dead" forms of CypA using CAN as a substrate, it becomes evident they're still active after all.
Night of the living "dead" enzymes
Panels B-G of Figure 3 in this paper directly show that every single one of the CypA mutants catalyzes CAN isomerization in solution (1). These spectra show peaks representing the chemical shifts of the nitrogen and hydrogen atoms of CAN's backbone amide groups in the presence of a small amount of CypA, so we are not looking at P90 directly. Fortunately, the chemical shift of the G89 amide is dependent on the isomerization state of P90. If the G89-P90 bond is in
trans, G89 shows up as the large peak at lower right in these panels, but if the bond is in
cis you get the small peak at upper left.
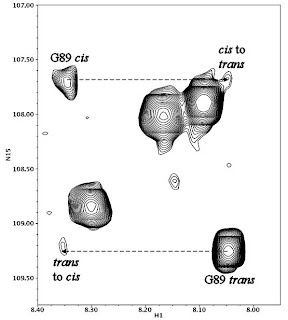
If you don't wait very long between determining the 15N chemical shift (y-axis) and the 1H chemical shift (x-axis), you get something that looks like panel A. If, however, you pause between determining the 15N shift and the 1H shift, you get cross-peaks representing the portion of CAN proteins that started the experiment in
trans and ended it in
cis, or vice-versa. The presence of these cross-peaks in the CAN/CypA samples, and their absence in the CAN-only sample (panel A), proves that catalysis is occuring. I've blown up the figure for H126A on the right to make things a little clearer. In this case the cross-peaks were pretty weak, but still in evidence.
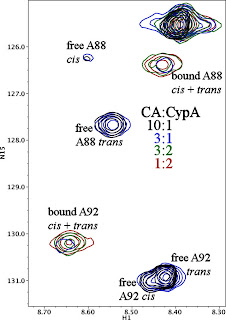
There was more variability when it came to
affinity, the strength with which CypA binds the CAN substrate. I've shown a complete titration for H126A on the left. As you can see, progressive addition of H126A causes the free CAN peaks to disappear while the new bound CAN peak grows in. This behavior is characteristic of
slow chemical exchange on the NMR timescale, and indicates a
high-affinity binding interaction. WT CypA binds CAN with a KD of 13 µM, and H126A probably has similar affinity. Note also that the bound state has a single peak for each residue, while the free state of CAN has separate
cis and
trans peaks. This indicates that the
cis and
trans isomers are interconverting rapidly on the enzyme, and constitutes additional evidence that H126A CypA is catalytically active.
This pattern was not repeated for all the mutants, however. H126A and W121Y had affinity similar to WT, while R55A, R55K, and F113W had significantly higher KD (lower affinity). You can see this clearly from the titrations in Figure 5. For each of these mutants, adding CypA to CAN caused the CAN peaks to move around in the spectrum, rather than disappearing and reappearing (R55K had a mixture of behaviors because the NMR timescale also depends on chemical shift). This peak shifting is characteristic of
fast chemical exchange on the NMR timescale and indicates relatively
low affinity. This wasn't the only change for those mutants.
Shifts in the rate-limiting step
An enzyme that doesn't have very high affinity for its substrate isn't necessarily in trouble. The NMR titrations of the R55A and R55K mutants indicate that their KDs are near 1 mM (Table 1). This is comparable to the affinity the WT protein has for the AAPF peptide, which gets catalyzed pretty efficiently. What does seem strange about this result is that the ZZ-exchange spectra are very similar.
The presence of single peaks for residues of CAN bound to WT suggests that the isomerization step is fast. Using relaxation-dispersion techniques, Daryl established that the net process rate (
kct+
ktc) for CAN on WT CypA was about 2200 /s. From the ZZ-exchange spectra we know that the total catalytic cycle goes a great deal slower (closer to 75 /s), from which we can deduce that isomerization is not the rate-limiting step. An analysis of the lineshapes suggested that the unbinding rate (
koff) was about 45 /s, which is close enough to the catalytic rate to indicate that this step is rate-limiting.
But if
koff is rate-limiting for this reaction, and the
koff for R55A and R55K is dramatically increased (as it must be, with lower affinity), we ought to be seeing a higher rate in the ZZ-exchange experiments, or maybe even not seeing independent cross-peaks at all. How can this reaction be going slowly enough to be seen by this technique? As it turns out, the titrations hold the key. When CAN is saturated with R55A CypA, you can clearly see independent
cis and
trans peaks
in the bound state (Figure 6, partially reproduced at right). This means that
cis-
trans interconversion
on the enzyme has gotten much slower.
In fact, the presence of those two peaks means that we can use the ZZ-exchange experiment
again, this time to determine the on-enzyme interconversion rate directly. The answer we get is about 20 /s, which is, within error, equivalent to the rate of the full cycle for this mutant. That means the rate-limiting step is no longer the unbinding of substrate, but rather
the isomerization step itself. There's only a minor change in overall catalytic efficiency, but this is the result of large changes in the rates of the individual steps that happen to cancel each other out.
Implications for the study of CypA-associated pathways
The best evidence available at the time supported the decisions Saphire
et al. made in setting up their experiment. Previous work had clearly shown that an H126Q mutation of CypA significantly reduces the protein's incorporation into virions (4,7). Saphire
et al. made an H126A mutation on this basis and seemingly assumed that the activity would be similar (6). Unfortunately, the evidence from the NMR spectra is that H126A binds to the capsid protein perfectly well and also catalyzes its isomerization. This does not
prove that the catalytic function of CypA is important for HIV-1 infectivity. However, on the basis of the existing experiments that possibility cannot yet be ruled out.
More broadly, these results demonstrate that claims about CypA's role in biology cannot be based on mutant studies alone. The mutants discussed here alter, rather than abolish, CypA's catalytic activity towards a biological substrate. Even those that appear not to bind the substrate in certain assays still display catalysis, because the strength of binding that is required for successful catalysis is considerably less than what is required for, say, a successful co-precipitation. The standard for assessing how a mutation has changed an enzyme's behavior needs to be one that pays attention to the various steps of the reaction and how changes in particular rates can compensate one another. Experiments that rely on
overexpression of a CypA mutant are particularly vulnerable to erroneous interpretation, because adding more enzyme is always an efficient way to compensate for a loss of activity and binding.
CypA and its related domains are very highly conserved across all vertebrates, yet its function was preserved even when apparently critical residues were dramatically altered by mutation. Our existing knowledge of protein sequences is limited to just a few examples from a relatively tiny number of species, and our structural and biochemical data encompass just a fraction of
that. Assessments based on these databases are likely to underestimate the functionally viable sequence space. Descriptions of function based on model systems are also suspect. That goes for
this system too the findings about CypA activity made with respect to CAN are not necessarily any more generalizable than the chymotrypsin assay results. CypA can bind an enormous number of potential targets, and what is true for one may not be true for another. Whenever possible, catalytic activity and binding affinity ought to be verified directly on the substrate of interest. Otherwise, you might find that an enzyme you thought was dead is still stumbling along.
References
1) Bosco, D.A., Eisenmesser, E.Z.,
Clarkson, M.W., Wolf-Watz, M., Labeikovsky, W., Millet, O., & Kern, D. (2010). "Dissecting the Microscopic Steps of the Cyclophilin A Enzymatic Cycle on the biological substrate HIV-capsid by NMR"
Journal of Molecular Biology DOI:
10.1016/j.jmb.2010.08.001
2) Bosco, D.A., Eisenmesser, E.Z., Pochapsky, S., Sunquist, W.I., and Kern, D. (2002) "Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A."
Proceedings of the National Academy of Sciences, 99(8), 5247-5252. DOI:
10.1073/pnas.082100499
3) Braaten, D., & Luban, J. (2001). "Cyclophilin A regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells"
The EMBO Journal, 20 (6), 1300-1309 DOI:
10.1093/emboj/20.6.1300 OPEN ACCESS
4) Dorfman, T., Weimann, A., Borsetti, A., Walsh, C.T., & Göttlinger, H.G. (1997). "Active-site residues of cyclophilin A are crucial for its incorporation into human immunodeficiency virus type 1 virions."
Journal of Virology, 71 (9), 7110-3 PMCID:
PMC192007 OPEN ACCESS
5) Gamble, T.R., Vajdos, F.F., Yoo, S., Worthylake, D.K., Houseweart, M., Sundquist, W.I., & Hill, C.P. (1996). "Crystal Structure of Human Cyclophilin A Bound to the Amino-Terminal Domain of HIV-1 Capsid"
Cell, 87 (7), 1285-1294 DOI:
10.1016/S0092-8674(00)81823-1 OPEN ACCESS
6) Saphire, A.C.S., Bobardt, M.D., & Gallay, P.A. (2002). "trans-Complementation Rescue of Cyclophilin A-Deficient Viruses Reveals that the Requirement for Cyclophilin A in Human Immunodeficiency Virus Type 1 Replication Is Independent of Its Isomerase Activity"
Journal of Virology, 76 (5), 2255-2262 DOI:
10.1128/jvi.76.5.2255-2262.2002 OPEN ACCESS
7) Zydowsky, L., Etzkorn, F., Chang, H., Ferguson, S., Stolz, L., Ho, S., & Walsh, C. (1992). "Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition"
Protein Science, 1 (9), 1092-1099 PMCID:
PMC2142182 OPEN ACCESS


Get complete info from
mwclarkson blog
















 Linear Mode
Linear Mode

