Why HisH doesn't fire until it sees the whites of PRFAR's eyes

The enzyme imidazole glycerophosphate synthase (IGPS) can be a bit of a lump. If you bind just one substrate it doesn't do anything, even though its two active sites are separated by more than 30 Å. Only if the second substrate
also binds does catalysis actually go at anything like a respectable rate. In a recent paper in
Structure researchers from Yale report evidence that this change of pace results from a change in dynamics.
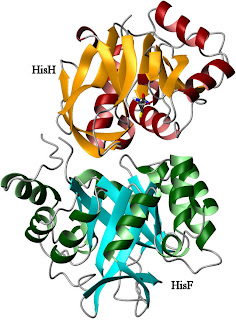
Apo- IGPS from
Thermatoga maritima
PDB code:
1GPWIGPS consists of two different protein subunits, HisH and HisF (right). HisH performs a relatively standard hydrolysis of glutamine, producing ammonia and glutamic acid. The ammonia molecule is then used by HisF as part of a cyclization reaction involving a weird nucleotide called PRFAR (with an IUPAC name that's just too long to bother with). The products of this reaction feed into the biosynthesis of histidine (as you might guess from the name) and the purines. In an example of poor planning, however, the active sites for these two reactions are separated by a great distance. Glutamine hydrolysis takes place near the interface between the proteins (which bind to each other with nM affinity), while PRFAR cyclization takes place at the far end of HisF (near the bottom of the image). This is too far for the ammonia to be efficiently transferred by any direct action of the enzyme itself. Therefore, the reaction proceeds when the NH3 travels down the
?-barrel of HisF to its distant active site (see image below left). The upside of this system is that ammonia gets where it needs to go. The downside of it is that unless the hydrolysis reaction only occurs when PRFAR is in position, this enzyme will be a little ammonia factory, costing the cell a fortune in nitrogen. Therefore, the cleavage reaction must be tightly regulated.
Enzymes can deal with this kind of demand in two ways. The first is to make the
binding of one substrate depend on another. This is called
K-type allostery because what is changed is the affinity (KD) of the enzyme for its substrates. Alternatively, the
rate of catalysis can be altered, which is called
V-type allostery because the velocity (
Vmax) of the reaction is changed. IGPS uses the latter approach. When glutamine binds, NH3 gets eliminated at a stately pace of about 10-3 /s. If PRFAR
also binds, however, HisH starts firing NH3 down the barrel at about 5 /s, which may not win many races but is a substantial enhancement. The question, then, is how the HisF active site lets the HisH active site know that PRFAR has arrived, when they are separated by more than 30 Å. Examining the enzyme complex in the presence of various ligands, James Lipchock and Pat Loria find evidence that changes to the dynamics of HisF are responsible for this communication.
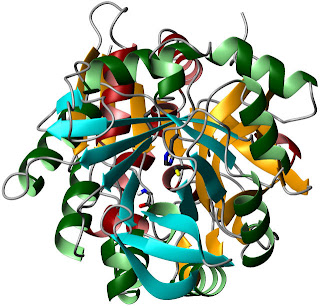
A rotated view, looking through the barrel
towards the HisH active site.The authors start by examining the energetics of PRFAR binding to IGPS. This event is endothermic, with an unfavorable enthalpy of binding. However, the entropic contribution is sufficiently large to overwhelm this effect. This could indicate a major increase in conformational entropy upon binding, or it could just be related to the behavior of water. Lipchock and Loria found that PRFAR binding to form the ternary complex had similar energetics. Of course, you can't form a ternary complex with actual substrates for very long, because catalysis would occur and change the affinities. They dealt with this using acivicin, a glutamine analogue that binds covalently to the active C84 of HisH.
Unfortunately, these thermodynamic data aren't particularly illuminating, so the authors proceeded with a high-resolution examination of the system. Because IGPS is a bit over 50 kDa in size, they chose to use methyl groups as their primary probes. Most of the remaining work in the paper uses ILV (
Isoleucine,
Leucine,
Valine) labeling, which takes advantage of the favorable relaxation properties of the methyl groups of those side chains.
Lipchock and Loria started by examining the enzyme in its
apo- state using relaxation-dispersion experiments. As I've mentioned before, these experiments detect exchange between different conformations on the microsecond to millisecond timescale. If this represents motion between two well-defined states, then the apparent relaxation rate at a given refocusing field strength will be a function of total process rate (
kex =
kab +
kba), the populations of the two states (pa and pb), and the chemical shift difference between them (??). If the exchange rate is fast on the NMR timescale (meaning that
kex >> ??), the last three parameters can be combined into a factor called
?ex.
This is how the authors fit their data, a choice they justified by stating that fitting the data to the full Carver-Richards formula (SI equations 8-18) gives similar answers for
kex but yields large errors in the populations and chemical shift differences. However, most of the dispersion curves look like data from slower exchange regimes. Unfortunately, I'm having trouble reconstructing their fitted curves from the parameters in any convincing way, in part because the equations in SI contain a few errors, so it's difficult to discuss where the vulnerabilities in this fitting procedure lie.
Using their approach, Lipchock and Loria find that only a few residues are experiencing conformational exchange, and they believe that the motions are primarily local. I'm not so certain on that point: a quick examination of SI Table 1 indicates that all but two methyls have
kex within error of 150 /s or so, which may indicate that most residues belong to a single process. However, most of the residues with similar fluctuation rates don't physically group in any obvious way (although V100 and V79 are adjacent).
Regardless of the particulars, it's clear that in the
apo- state, few of the methyl groups in HisF are experiencing any kind of µs - ms fluctuation. Binding of acivicin to HisH doesn't change this too much. Within the bounds of the fitted error, the extracted dynamics parameters are the same for many residues. The exceptions are the adjacent residues V79 and V100, and L153?1, which has an odd halving of both rate and the combined parameter.
Also, as you can see in SI Table 2, the
R2° values in this state are significantly lower than
apo- IGPS. This is difficult to interpret without knowing exactly how the experiment was performed; they could represent additional ns fluctuations, the removal of some very fast global process, or simply different deuteration efficiency. However, some methyls do not appear to have large changes in their
R2° values (e.g. V56?2, I73?1, L94?1). Most of the spurious factors that would give rise to the observed changes in
R2° should affect all residues more or less equally; the lack of uniformity suggests this may be worth following up on.
When Lipchock and Loria added PRFAR to the system, all hell broke loose. Many of the amide groups in the protein had their signals broadened beyond the detection limit, indicating conformational exchange on the intermediate timescale. In addition, a large number of methyl groups showed evidence of conformational exchange.
Here the fluctuation is obviously a genuinely incoherent one. Not only do the fitted
kex values vary wildly across the protein, they also have poor fitting characteristics (including fitted errors greater than 100%), and enormous differences between adjacent methyls on a side-chain (
e.g. L153?1,2). This suggests that the two-state model might be inappropriate, which is what you would expect for widespread and incoherent fluctuations among contiguous residues. For atoms that are in close proximity, a two-state exchange model presupposes some kind of coherent fluctuation, because in a chaotically fluctuating system, well-defined, relatively long-lived states don't exist.
Of course, in the absence of well-defined, relatively long-lived states, it's difficult to understand what all this motion
does. It's therefore very interesting that when acivicin
and PRFAR are bound to the enzymes, forming the ternary complex, all the methyls can be fit to a single conformational exchange process with a rate of about 225 /s. That is, the formation of the ternary complex causes the dynamics to become a global (or nearly so), coherent process.
So, what does all that wiggling accomplish? Lipchock and Loria point out that in the apo- structure of HisH, the backbone amide group of V51 is improperly positioned. Its role in the reaction is to stabilize the negatively charged oxygen in the tetrahedral intermediate of the reaction. However, as you can see in their Figure 9, this amide points away from the reactive cysteine in the
apo- state. In order to fulfill its function, this loop must rotate about 180° from the
apo- position.
The authors hypothesize that the coherent fluctuations of HisF in the ternary complex are transmitted to the active site of HisH and make it possible for this rotation to occur. Consistent with this model, the binding of PRFAR to HisF causes the amide resonance of G50 to broaden out due to chemical exchange. The titration (in Figure 9) looks a little strange; it's not clear why the peak shifts between 4% and 20% saturation, or why no points are shown from 33% to 100%. While neither glutamine nor acivicin was bound in this experiment, it at least confirms that information about PRFAR binding to HisF can reach the binding site of HisH as changes in dynamics.
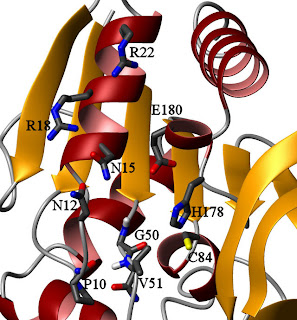
HisH active site, looking up the barrel from HisFThis might seem like an odd mechanism, because this particular loop in HisH has no points of close contact with HisF in the crystal structure. By contrast, there appear to be several points of contact between HisF and the region around catalytic triad members H178 and E180, so one could argue that they are more likely responsible for the observed effect. In the
apo- state, however, the backbone amide of V51 is hydrogen-bonded to the carbonyl oxygen of P10 (see figure on the right). Fluctuations in that loop, perhaps transmitted from HisF through contacts to HisH residues N12, N15, R18, and R22, could destabilize that bond and encourage rotation. The HisF residues I93 and I73, which are part of the dynamic network in the ternary complex, lie in this region. However, the bulk of the contacts are to the backbone, and alanine dynamics (reflective of main-chain motions) do not appear to have been studied in the ternary complex. A good look at HisF A70, A89, and A97 when both ligands are bound may give some insight into whether this is the transmission point, and some data on the ILV residues of HisH in this region would also help examine this hypothesis. It might also be valuable to mutate P10 to something more flexible to see if the regulation is altered.
The authors point out that the fluctuation rate is many times larger than
kcat. The relevant rate, however, is the rate at which the complex enters the catalytically-competent state, which is probably much lower than the total
kex. Here, a fit to the full Carver-Richards equation yielding populations would have been enormously valuable. It's therefore possible (but not yet proven) that the HisF fluctuations are rate-limiting for HisH catalysis, which would after all be an easy way to achieve V-type regulation.
This is another case in which dynamics allow a protein to reconcile incompatible functional requirements. IGPS must be nearly inactive in the absence of PRFAR, yet still achieve a significant rate enhancement in its presence. Although much work remains to confirm the hypothesis that the dynamics are solely responsible, it appears that fluctuations in HisF may enable HisH to adopt an alternate conformation that is catalytically competent while generally favoring the inactive structure.
Lipchock, J., & Loria, J. (2010). "Nanometer Propagation of Millisecond Motions in V-Type Allostery"
Structure, 18 (12), 1596-1607 DOI:
10.1016/j.str.2010.09.020

Get complete info from
mwclarkson blog















 Linear Mode
Linear Mode

