Smaller NMR files
Background
One important issue we had noticed with Mnova NMR files is that they can be quite large, particularly when a document contains several 2D spectra. At first sight, file size should not be a big concern, especially considering the large storage capabilities available today, either locally (i.e. hard disks with sizes in the order of Terabytes) or in the cloud (Dropbox, Google Drive, Skydrive, etc).
On the other hand, the tremendous advancements on both technological and methodological fronts have made possible the acquisition of enormous volumes of data. For example, IBM has estimated that 2.5 quintibillion bytes of data are being generated each day, with more than 90 per cent of which created in the last two years. Whilst it is difficult to scale this level of information into analytical data (i.e. NMR spectra), it is quite likely that they also follow a similar growth.
At Mestrelab we have devoted major efforts to the development of new technologies which would allow Mnova to reduce the size of NMR spectra while preserving their informational content. This will be elaborated in the following section.
Lossless and lossy compression
Roughly speaking, there are two two different classes of compression methods:
lossless and
lossy.
Lossless techniques allow the data to be compressed, then decompressed back to its original state without any loss of data. Well-known algorithms for this type of compression are Zip and Rar methods. Compression rates for lossless techniques vary but are typically around 2:1 to 3:1, e.g in medical images. In the particular case of high resolution NMR spectra, there re some relevant characteristics that diminish the performance of this type of algorithms.NMR spectra consist mostly of a noisy background and hence appear as essentially random numbers to the algorithm which makes lossless compression rather ineffective; in general, NMR spectra can be compressed by no more than 10-30% (on average) using lossless compression schemes.
Lossy techniques do not allow the exact recovery of the original data once it has been compressed, but this loss of information can be modulated in such a way that it can be virtually negligible. In the particular case of NMR, we have applied several advanced compression techniques [1, 2] which afford extraordinarily high compression rates while preserving all the spectral information. In some cases, compression rates in the order of 800:1 can be achieved, although for practical uses and in order to avoid any potential loss of information, more moderate rates are recommended.
An example
In the figure below, the DQF-COSY of Taxol (Paclitaxel) is shown at its original uncompressed format (left) and after being compressed 100 times with the new built-in compression algorithm in Mnova and decompressed back (right). Both spectra have been displayed with the same contour levels. Can you spot the differences?
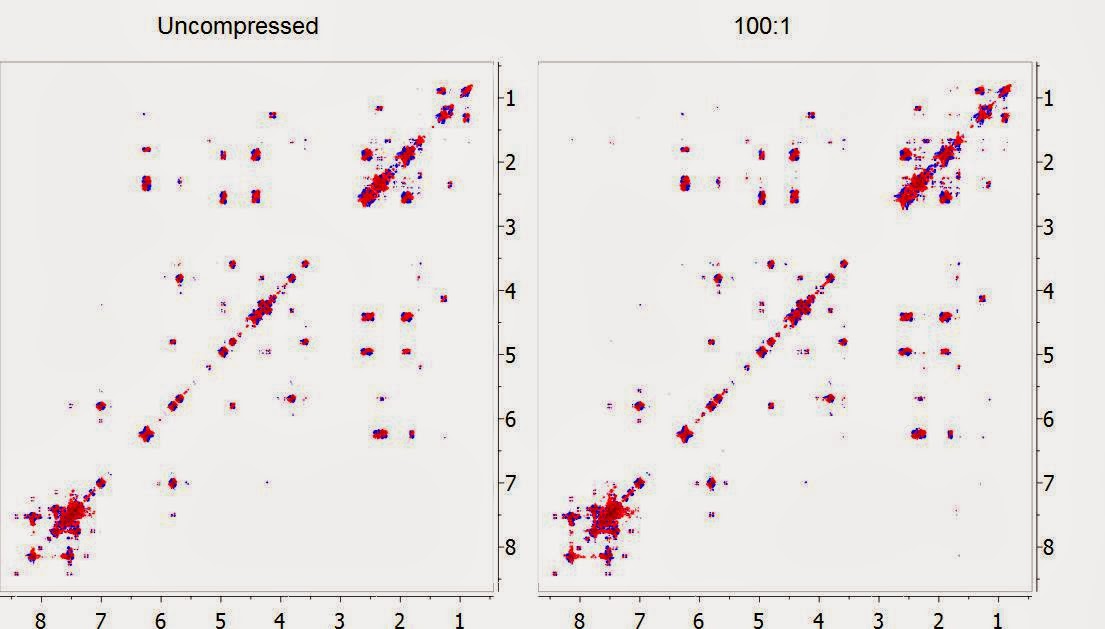
Whilst we have done lots of numerical tests to make sure that at this high level of compression all the spectral information is preserved (see [1] and [2] for more details), a simple yet intuitive way to visualize whether the compression has been effective is by subtracting the uncompressed spectrum with the compressed counterpart. In this example, this is the residual spectrum:
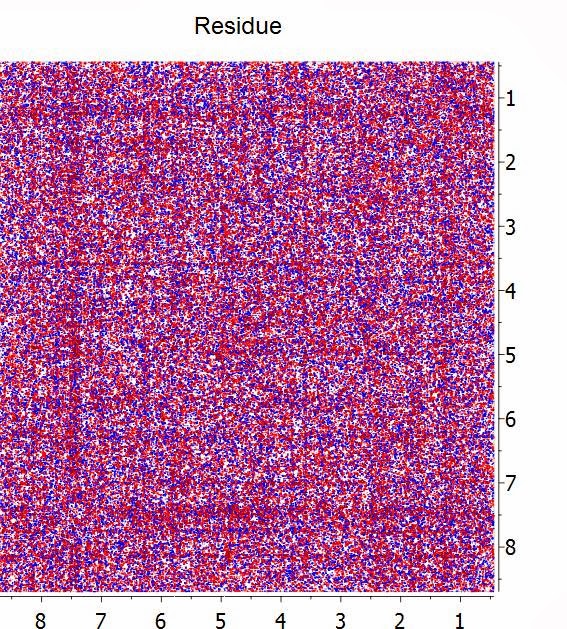
Basically, all that it remains is noise and no structures (cross peaks) are visible on the residual.
A practical guide with Mnova 9.0
This is how compression works in Mnova NMR. First, all the compression options are available in the global Preferences of the software (command
Edit / Preferences), in the
NMR/Save page (see below):
At this point, there are two different compression mechanisms:
FID compression: The FID is the most important component of an NMR spectra where all the actual recorded information is stored. We dont want to miss even a single bit of this data and hence, the FID is only compressed using a lossless algorithm. Of course, the compression ratio will be much more modest, but it is critical to preserve all this information.
FT spectrum Compression: This is where the lossy compression algorithm can be applied, in the frequency domain spectrum. Actually, it is also possible to use a lossless algorithm but in order to achieve high compression ratios, the lossy method should be selected. Whilst values of 100:1 or even higher should give good results, it would be more sensible to use more moderate values, in the range of 10:1 20:1.
Final notes
The fact that Mnova NMR documents keep both the original recorded FID (which can optionally be compressed using the lossless technique) as well as the processed NMR spectrum (which can optionally be compressed using the lossy technique) explains why the resulting compressed document is not as small as one could expect after having compressed the data with high compression ratios. The FID might contribute significantly to the final file size. Of course, the differences will be more appreciated in 2D NMR spectra processed with Zero Filling or Linear Prediction so that the final data matrix becomes significantly larger than the time domain vectors.
On the other hand and considering again the point that Mnova always keeps a copy of the original FID, why we dont just save this FID plus the processing commands required to reconstruct the processed spectrum as other NMR applications do? Actually, this is a nice approach (under some circumstances) and would yield the best compression ratio achievable. Unfortunately, this does not work well for many applications and introduce some additional difficulties. Just to give a simple example: You have processed a 2D spectrum which was acquired with a NUS scheme and you have applied some additional time-consuming analysis operations (i.e. 2D-GSD based peak picking). In this particular case, opening this single spectrum would take several seconds (if not minutes). Having the ability to access directly to the processed spectrum without the need to reprocess it may be very handy.
References:
[1] Carlos Cobas, Pablo G. Tahoces, Manuel Martin-Pastor, Mónica Penedo, F. Javier Sardina (2004), Wavelet-based ultra-high compression of multidimensional NMR data sets, J. Magn. Reson. 168: Pages 288295.
DOI:
http://dx.doi.org/10.1016/j.jmr.2004.03.016
[2] C. Cobas, P. G. Tahoces, I. Iglesias Fernández (2008), Compression of high resolution 1D and 2D NMR data sets using JPEG2000, Chemometrics and Intelligent Laboratory Systems, 91, 141-150
DOI::
http://dx.doi.org/10.1016/j.chemolab.2007.10.009
More...
Source:
NMR-analysis blog















 Linear Mode
Linear Mode

