A Novel qNMR Technique: Quantitative Global Spectrum Deconvolution (qGSD)
Ever since chemists meddled (successfully) into NMR, with the pioneer work made by
Proctor and
Yu [1] more than 67 years ago, it was implicitly used as a
quantitative technique. Indeed, from the very early days of NMR, it was found that the intensity (or area) of the NMR signals (under proper operating conditions) was proportional to the number of nuclides contributing to it. Already in 1953, Jarrett, Sadler, and Shoolery [2] showed the excellent precision of NMR (CW at that time) for the quantitative analysis of a tautomeric mixture, despite the limited measurement resources (see Figure below).
 Figure 1
Figure 1. 1H-NMR spectrum from Reference [2]
Interestingly, quantitative analysis by NMR (qNMR from now on) is nowadays experiencing a kind of Renaissance with many applications in various fields, such as pharmaceutical and food sciences, manufacturing of reference materials, or metabolite determination in human body fluids. Furthermore, it is now being considered as an official analytical method for purity determination or assay of concentration of organic compounds.
From sum integration to qGSD
One of the key factors for the successful application of qNMR is the method used to estimate the peak integrals in the spectrum. This has been, and still is, largely dominated by the so-called
sum integration in which the computer simply calculates the sum of all data points within a spectral region. It is well known that this method is very sensitive to phase or baseline distortions, but we can also assume that those spectral artefacts can be properly corrected (even fully automatically) by the NMR software.
The main problem of this method is its
inability to deal with overlapping peaks. This is a particularly serious problem when an "external" signal overlaps within the area of interest. This signal can be solvent, it can come from another compound or even from the same compound itself.
This problem is illustrated in the 1H-NMR spectrum of Santonine (Figure 2).
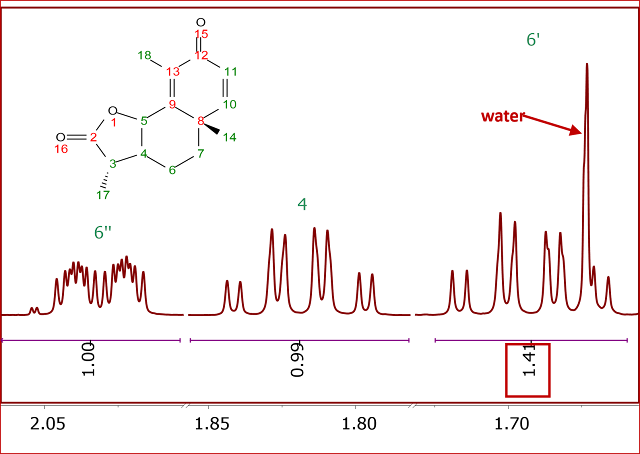 Figure 2
Figure 2. 1H NMR spectrum of Santonine. Integrals calculated using standard sum method
Notice the multiplet at ~1.7 ppm that is contaminated with residual water peaks and therefore, the integreal corresponding to the proton 6 is
overestimated by a significant amount.
The contribution of that solvent peak could be removed by different methods, ranging from acquisition (e.g. pulse sequences for solvent suppression), post-processing or deconvolution techniques or a combination of the three.
Global Spectral Deconvolution (GSD) is a powerful alternative to the standard integration method providing a number of advantages. First, it is pretty
insensitive to baseline distortions, although it requires the spectrum to be phase corrected. Most importantly, it can deal very efficiently with the
problem of peaks overlap as it can be seen using the same example of Santonine used before.
(Figure 3)
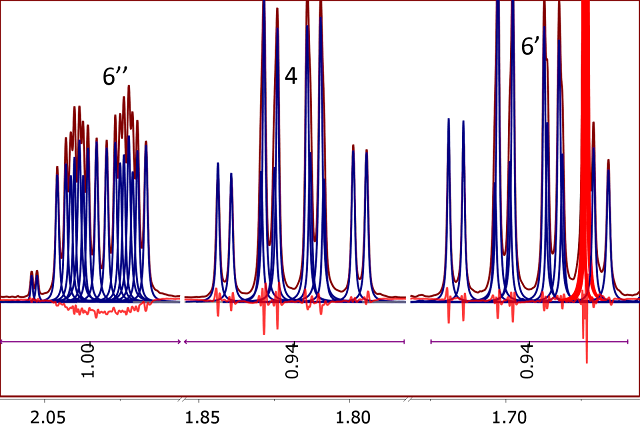 Figure 3
Figure 3. 1H NMR spectrum of Santonine. Integrals calculated using GSD
Compared to the classical point-to-point integration, GSD yields better relative integrals, but the quality of the fit is not optimal (notice the residues under the spectrum). Nevertheless, they are generally of sufficient quality to be used all but the most demanding quantitation, It has been at the core of Mnova data analysis for many years, and has proved to be highly reliable and computationally efficient. However, whilst GSD has been designed to be used for problems involving automatic spectral analysis such us those required to perform molecular structure characterization, it was not intended for very accurate quantitative analysis (e.g. qNMR).
GSD uses a Generalized Lorentzian function to model the experimental lineshapes. This is a very flexible model that covers a broad range of shape variations for NMR resonances (Figure 4).
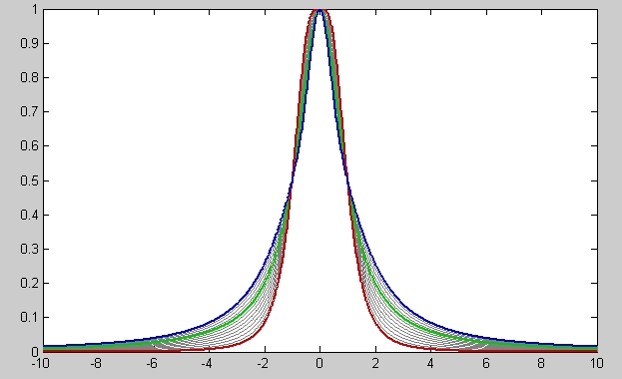 Figure 4
Figure 4. The shape parameter in this graph ranges from 1.00 (blue) to +2.00 (red). For 0.00 (green), the line is a perfect Lorentzian. Red line is pure Gaussian and the gray lines are generalized Lorentzians.
The quality of the quantitation depends, amongst other factors, on how well theoretical models describe experimental NMR resonances. Careful analysis shows that even a flexible model like the Generalized Lorentzian cannot precisely fit all possible experimental lineshapes, as evidenced by non-zero fitting residual in Figure 3.
In order to overcome some of the limitations of GSD, we have now introduced qGSD (quantitative GSD), available in Mnova 12. It is based on careful analysis of the residuals after GSD processing, and correcting GSD lineshapes in a way which minimizes the residuals. In Figure 5 we can see that now the residues are significantly smaller compared to the results obtained with plain GSD.
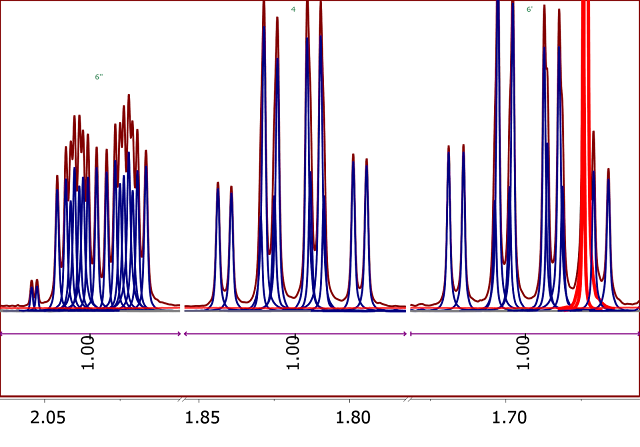 Figure 5
Figure 5. 1H NMR spectrum of Santonine. Integrals calculated using qGSD
Using qGSD with Mnova 12
In order to enable qGSD, it is necessary to access to the Peak Picking options in Mnova making sure that GSD method is selected (see Figure 6).
 Figure 6
Figure 6. Enabling qGSD in Mnova
Next, under the Advanced options, qGSD check box must be ticked and an appropriate number of Improvement cycles selected. A number between 5 and 10 usually gives good results. However, it is important to bear in mind that qGSD is computationally intensive: can take a couple of minutes, especially with a large number of cycles.
For this reason, it is recommended to use qGSD only when highly accurate integrals are needed. For other applications, such as multiplet analysis, or structure confirmation, plain GSD would be a more efficient method.
qGSD with poorly shimmed spectra
To illustrate the lineshape flexibility offered by qGSD, a poorly shimmed spectrum has been used with GSD and qGSD (Figure 7). It is clear that qGSD does a much better job. That said, this should not not be a replacement for a properly acquired spectrum.
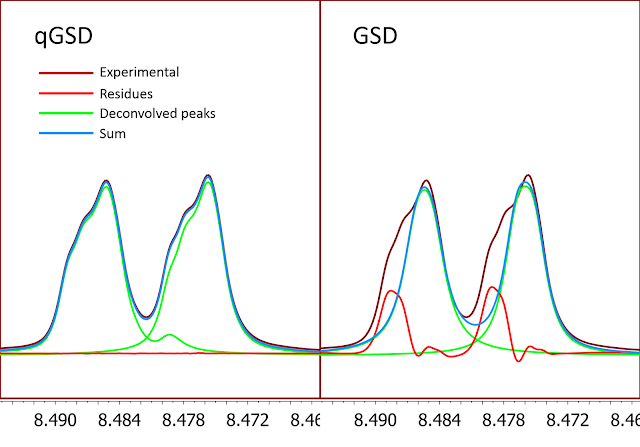 Figure 7
Figure 7. qGSD vs GSD on a poorly shimmed spectrum
Conclusions
qGSD (quantitative Global Spectral Deconvolution) represents Mnovas latest innovation that combines the power of deconvolution techniques to handle overlapped signals with the robustness of sum integration of isolated resonances but with the ability to deal efficiently with overlapped peaks.
Our preliminary tests demonstrate that qGSD is able to provide accuracy which is supperior to the sum integration even when there is signal overlap
References
[1] W. G. Proctor, E C.Yu, Phys. Rev 77,717 (1950)
[2] H. S. Jarrett, M. S. Sadler, and J. N. Shoolery, J. Chem. Phys., 1953, 21, 2092.
More...
Source:
NMR-analysis blog



















 Linear Mode
Linear Mode

