Alternative side-chain structures from methyl CPMG

As I have mentioned before on this blog, the use of tools like CS-ROSETTA holds the promise of
determining protein structures using only the chemical shifts of its backbone atoms. In addition to potentially making NOEs and RDCs redundant, this technology allows biologists to determine the conformations of minor members of the structural ensemble, which are very difficult to obtain using conventional approaches in population-dominated techniques like NMR and X-ray crystallography. There are two limitations here, however. First, we only gain insight into the backbone, and as we know,
the positions of side chains in minor states can be critical for function. In addition, backbone chemical shifts are not always available due to relaxation problems. Both weaknesses could, in principle, be addressed by extracting conformational information from the chemical shifts of methyl groups, which report on side-chain behavior and continue to give good signal even in very large proteins. This is the rationale behind a series of recent papers from the Kay lab [1-3] intended to determine changes in side-chain rotameric state from methyl relaxation-dispersion data.
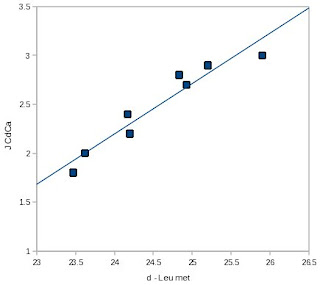
The roots of this idea have been around for a while, dating back at least to a 1996 paper in
J. Biomol. NMR [4]. I've reproduced one of MacKenzie
et al.'s figures at right, and as you can see, for this protein (a peptide of glycophorin A), the correlation between the C? chemical shift and
JC?C? is quite striking. However, the quality of the correlation appeared to be protein-dependent, as the R2 for this relationship was significantly lower for staphylococcal nuclease side-chains, possibly because they were positioned in a less homogeneous chemical environment than a lipid bilayer.
A more systematic study was recently performed by Bob London and co-workers from the National Institute of Environmental Health Sciences [5]. They extensively compared side-chain rotameric angles extracted from the
PDB to side-chain chemical shift data from the
Biological Magnetic Resonance data Bank to see what correlations emerged. They expected to see that the chemical shifts of the carbons depended on the side-chain dihedral angles due to the "?-substituent effect", which is believed to alter chemical shifts due to bond polarization caused by steric interactions. Although there are some complications due to other effects, this prediction turned out to be true, broadly speaking.
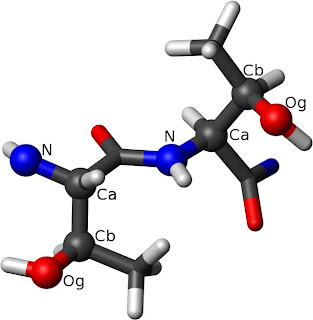
The left Thr has ?1=-60° while the right one has
?1=60°. The rotation around the C?-C? bond from
N to O? defines the dihedral angle.London
et al. found clear correlations between chemical shift and rotameric state for threonine, for instance, which has a true chiral center at C?. For ?1 of ± 60° (these angles are also referred to as
gauche±), the chemical shift of the methyl carbon was around 22 ppm, while for ?1 of 180° (also called
trans) the chemical shifts cluster loosely around 19 ppm. More broadly, London
et al. observed that sterically crowded rotamers tended to move aliphatic carbon chemical shifts upfield. Structurally, the difference between these dihedral angles is that in the ±60° positions, C?2 has steric interactions with only one heavy atom (
i.e. the amide N or carbonyl C), while in the 180° position it interacts with two.
As one might expect given the results of Mackenzie
et al., London
et al. also found a straightforward relationship in the case of the leucine ? carbons, where the population of rotamers could be determined rather simply using the difference between the ?1 and ?2 chemical shifts. While this only specifically gives the population of the
trans rotamer (where C?1 is on the opposite side of the C?C? bond from C?), it turns out that, due to unfavorable sterics, population of the
gauche- conformation is vanishingly small in the PDB, so that one can assert with some confidence that everything not in
trans is in
gauche+. Also, London
et al. noted that the ?1 and ?2 angles were highly correlated for leucines, so that in principle the entire side-chain conformation could be defined using just the difference in C? chemical shifts.
Hansen
et al. [1] decided to use the chemical shift-rotamer relationship to analyze the minor conformations of leucines in mutants of the Fyn SH3 domain. The G48M mutant is in a rapid equilibrium between folded and unfolded forms, while the A39V/N53P/V55L triple mutant appears to primarily exchange to an intermediate state. Using a combination of CPMG-based relaxation-dispersion experiements and HSQC/HMQC, the Kay lab were able to determine the chemical shifts of the leucine methyls in the alternate state for each mutant, and thus derive populations for the
trans rotamer. In the unfolded state, on expects to see ~60-70% population of the
trans rotamer. The folded state of Fyn SH3 has several leucines that lie outside this range, but in the minor form of G48M nearly all of them lie within it, consistent with the existing finding that this state is unfolded. In the case of the triple mutant, some leucines move into the unfolded range in the minor state, while others remain outside of it. This is consistent with the assignment of the minor state as a partially-folded intermediate.
In a subsequent paper, Hansen
et al. derived a relatively simple method for estimating the population of the
gauche- rotamer state for the isoleucine ? carbon and applied it to the same system [2]. The situation for the Ile C? is somewhat more complicated than that of leucine. Because it is an isolated methyl group, and the rest of the side chain has a complicated topology, as many as four unique rotamer positions are distinctly populated in the PDB. However, in solution only the
trans and
gauche- configurations are expected to be significantly populated.
The Fyn SH3 domain has two Ile residues, which by this technique appear to be populated primarily in the
gauche- rotamer (I28) and the
trans rotamer (I50) respectively. In the intermediate state (results from the unfolded state are not reported) both isoleucines populate the
gauche- rotamer to about 20%. The authors interpret this as a non-native interaction in the case of I28 and a slight increase in dynamics in the case of I50. However, it seems that these values could also support a case that both side chains are totally (or almost totally) solvent-exposed in the intermediate state, and thus adopting random-coil configurations.
One might also take issue with the idea that an increase from 0 to 20% of an alternate rotamer population represents a "slight" increase in dynamics. It's difficult to make any firm statement in this regard because we don't actually know the rotamer distribution in the folded state: C?1 may be entirely in
trans, or averaged somehow between all of the non-
gauche states. The authors take the folded state to be essentially pure
trans, from which one would plausibly expect to observe an
order parameter of 0.8 or higher for the methyl group (according to the rough calculations in [6], see reproduced figure on left). Based on the population, the order parameter would decrease to around 0.5 in the intermediate, a fairly large change.
However, this does not undermine the conclusion that the core is relatively well-formed in the intermediate. One perplexing feature of methyl side-chain order parameters is that they correlate poorly with nearly every structural feature one might expect to explain them [7]. Solvent-accessible surface area, packing density, and depth of burial are all rather poor predictors of side-chain dynamics. By the same token, more rudimentary measures, such as methyl distance from the backbone, are relatively robust predictors of dynamics, even though they ignore the higher-order structure of the protein. The upshot of this is that any data obtained about the dynamics of side-chains in minor states will need to be interpreted conservatively.
In the most recent offshoot of this research, Hansen and Kay published a paper correlating the chemical shift of valine C? methyls with the rotameric state [3]. Unfortunately, this is not a case where there's a simple calculation that can accurately spit out the ?1 angle, and because of the ?-branched structure of the amino acid, it's not possible to rule out one of the possible angles
a priori. As their Figure 2 shows, the relationship between the chemical shifts and the rotamer is complicated and may also vary with the local secondary structure. Instead of a simple formula, they were able to derive a "surface" reflecting probabilities of particular rotamer arrangements based on the shifts, which can then be analyzed using a program they wrote. They subsequently validated this approach on a very large protein complex, the half-proteasome, by comparing the chemical shift-derived rotameric states to those observed in crystallographic data.
I tested their chemical-shift based predictions against some of my own data (a web-based version of the program is available at
Flemming Hansen's website) and wasn't exactly blown away by the results. Of 10 methyls in my protein, the primary rotamer was completely wrong (as determined by experimental 3
J measurements) in two cases, and the population of the primary rotamer was dramatically overestimated in another two. However, the two side-chains with incorrect rotamer determinations were both adjacent to tryptophan side-chains, and in those cases the ring currents may have altered the chemical shift enough to inferfere with the calculation. Because aromatic rings are likely to be present in the core and may enhance the possibility of observing chemical exchange, this may bear further investigation. Nonetheless, the primary rotamer was usually correct chosen, and so these calculations can serve as at least a starting point for structural analysis.
These introductory studies are fairly encouraging, and suggest that it should be possible to use CPMG experiments to assess structural features of minor states beyond just the backbone conformation, even in very large systems. This may be especially helpful in analyzing the dynamics of proteins with hydrophobic active or regulatory sites. As hydrophobic surfaces are often involved in protein-protein interactions, an improved understanding of these critical binding events may result.
Disclosure: I have co-authored a paper with Bob London's group, as well as several (obviously) with Andrew Lee's.
1. Hansen, D., Neudecker, P., Vallurupalli, P., Mulder, F.A.A., & Kay, L. (2010). "Determination of Leu Side-Chain Conformations in Excited Protein States by NMR Relaxation Dispersion."
Journal of the American Chemical Society, 132 (1), 42-43 DOI:
10.1021/ja909294n
2. Hansen, D.F., Neudecker, P., & Kay, L.E. (2010). "Determination of Isoleucine Side-Chain Conformations in Ground and Excited States of Proteins from Chemical Shifts."
Journal of the American Chemical Society, 132 (22), 7589-7591 DOI:
10.1021/ja102090z
3. Hansen, D.F., & Kay, L.E. (2011). "Determining Valine Side-Chain Rotamer Conformations in Proteins from Methyl 13C Chemical Shifts: Application to the 360 kDa Half-Proteasome."
Journal of the American Chemical Society, 133 (21), 8272-8281 DOI:
10.1021/ja2014532
4. MacKenzie KR, Prestegard JH, & Engelman DM (1996). "Leucine side-chain rotamers in a glycophorin A transmembrane peptide as revealed by three-bond carbon-carbon couplings and 13C chemical shifts."
Journal of Biomolecular NMR, 7 (3), 256-60 PMID:
8785502
5. London, R., Wingad, B., & Mueller, G. (2008). "Dependence of Amino Acid Side Chain 13C Shifts on Dihedral Angle: Application to Conformational Analysis."
Journal of the American Chemical Society, 130 (33), 11097-11105 DOI:
10.1021/ja802729t
6. Hu, H., Hermans, J., & Lee, A. (2005). "Relating side-chain mobility in proteins to rotameric transitions: Insights from molecular dynamics simulations and NMR"
Journal of Biomolecular NMR, 32 (2), 151-162 DOI:
10.1007/s10858-005-5366-0
7. Igumenova, T., Frederick, K., & Wand, A. (2006). "Characterization of the Fast Dynamics of Protein Amino Acid Side Chains Using NMR Relaxation in Solution."
Chemical Reviews, 106 (5), 1672-1699 DOI:
10.1021/cr040422h


Get complete info from
mwclarkson blog















 Linear Mode
Linear Mode

